Huntington’s disease is a hereditary, incurable brain disorder that damages brain cells. There are many effects of depression, including effects on movement, thinking and mood.
Genetic mutations cause toxic proteins to collect in the brain, causing Huntington’s disease.
An estimated 3–7 in every 100,000 Western Europeans suffer from the disease. Japanese, Chinese, and African descendants are less likely to have it, according to Genetics Home Reference.
People aged 30–50T usually exhibit the first signs.
Read: Focal Dystonia
What is Huntington’s disease?
Huntington’s disease is a neurological disorder. It is caused by a gene mutation, which results in inherited disease. Proteins that are toxic accumulate in the brain and lead to neurological damage.
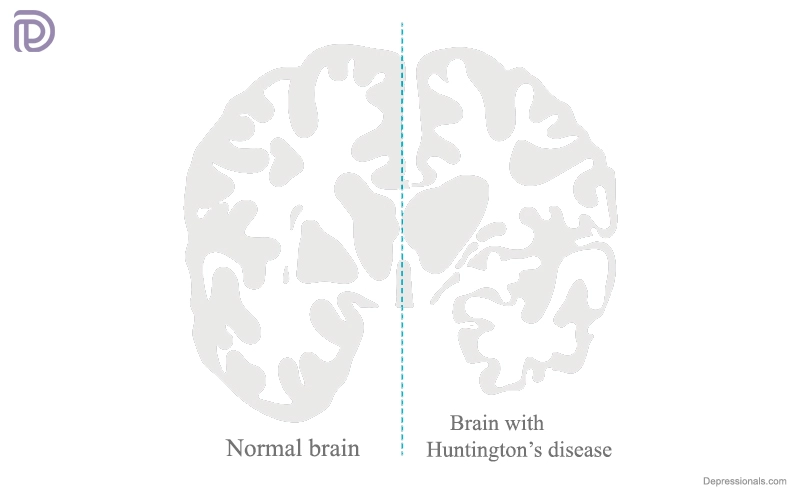
Different brain sections are affected by the disease, which affects movement, cognition and behavior. People with the disease find it difficult to walk, talk, reason, swallow and think. Eventually, full-time care will be required. There is a risk of death from the condition.
Treatment is currently unavailable, but symptoms can be relieved.
Symptoms of Huntington’s
Most Huntington disease symptoms and signs appear in adults aged 30-50 but may also appear in younger adults.
Here are some key symptoms:
- Mood and personality changes
- Depression
- Difficulties with memory, reasoning and judgment
- Inability to control movements and movements
- Speech and swallowing problems
Symptoms may vary from individual to individual. People experiencing depression may notice changes in their motor skills first. They may also exhibit unusual behavior or moods.
Read: Generalized Dystonia
Early signs and symptoms
If no member of the person’s family has ever been diagnosed with Huntington’s disease, a physician may not recognize the symptoms at an early stage. Getting a diagnosis can take time.
There are several symptoms that may indicate Huntington’s disease:
- Uncontrollable movements
- Along with clumsiness and lack of coordination
- Stumbling
- Slight changes in mood and emotions
- Difficulty concentrating and working
- Temporary memory loss
- Depression
- Irritability
Additionally, the person may appear lethargic and lacking in initiative, losing motivation and focus.
You may also lose things and lose a person’s name as early signs of Huntington’s disease. It is not uncommon for people to do these things from time to time. An individual with Huntington’s disease experiences these symptoms gradually becoming more and more severe.
The middle and later stages
Throughout the progression of the disease, there is an increase in physical symptoms, motion control loss, as well as emotional and cognitive changes.
Read: Chronic Fatigue Syndrome
Physical changes
You may notice the following changes:
- Speaking problems, including difficulty finding words and slurring
- Weakness caused by weight loss
- Loss of function in the diaphragm and mouth muscles can create difficulties with eating and swallowing
- An increased risk of choking develops with advancing age
Uncontrollable movements of the body include:
- Facial movements
- Jerking movements of the head and face
- Arms, legs, and body movements that are twitchy or flickering
- Stumbling and lurching
Movements that are uncontrollable become more frequent and more intense with time. Eventually, their speed slows down as muscles stiffen.
Emotional changes
Huntington’s syndrome produces emotional symptoms as well as psychological symptoms.
There are two major categories of mental illness: those that affect people with and without the disease. The chances of an individual with Huntington’s developing delusions, mania, or obsessive-compulsive disorders are approximately 40%.
Huntington’s affects two categories of mental state changes. The first involves changes caused by changes in the brain itself. Examples of such changes are:
- Aggression
- Anger
- Antisocial behavior
- Apathy
- Depression
- Excitement
- Frustration
- An increasing lack of emotion
- Moodiness
- Stubbornness
- Cognitive changes
Other possible factors include:
- An inability to take initiative
- Reduced organizational skills
- Disorientation
- Difficulty focusing
- Problems with multitasking
Due to cognitive changes, including disinhibition and impulsivity, Huntington’s disease sufferers face a suicide risk up to 10 times the national average. Those who are close to individuals with this condition should be alert to the signs of suicide ideation.
Read: Mixed Dementia
The later stage
The person will eventually be incapable of walking or talking and will require nursing care to live.
The majority of what they hear will be understood by them, and they will be aware of family and friends around them.
Complications
Frustration and depression can occur when a person has difficulty doing things that were once easy.
A patient’s illness may be exacerbated if they lose weight, as it weakens the immune system and makes them more prone to infections.
A person with Huntington’s disease usually does not die of the disease, but can become choking, pneumonia, or infected with another illness that may be fatal.
Huntington disease cause
An abnormality in chromosome number 4 causes Huntington’s disease.
This gene typically produces a protein called huntingtin (HTT). However, the mutation makes the gene larger than it ought to be. CAG, the three building blocks of DNA, are produced in excess as a result. It is normal for CAG to repeat 36 times or less, but when it occurs in Huntington’s disease, it may repeat 36 times or more.
HTT proteins become toxic when this type of change occurs. Over time, the toxic protein begins to damage certain brain cells in the brain as it accumulates.
People with repetition between 36 and 39 are at a greater risk of developing Huntington’s disease. An individual will almost certainly develop the condition if it occurs 40 times or more.
Read: Vascular Dementia
Where does it come from?
The condition is inherited through the autosomal dominant gene. Therefore, it is possible to inherit the mutation from one parent only, and not both.
One copy of the gene is normal and the other is mutated in a person with this gene mutation. The mutated copy or the typical copy is inherited by the offspring. He or she won’t get Huntington’s disease if they inherit the typical copy. If they inherit the mutant copy, they will.
The odds are 50% for each child to inherit the mutation. They’ll each have a 50% chance of passing it on to their kids if they inherit the mutation. It may affect more than one generation.
Someone who doesn’t inherit the gene mutation won’t get sick and can’t pass it on to their kids. If they reach the age when symptoms should appear, a child who inherits the mutation will get Huntington’s. Symptoms appear up to 10% of the time before the 20th birthday and 4–11% after the 60th.
Huntington disease diagnosis
The doctor will conduct an examination and ask about the person’s family history, medical history, and recent emotional changes to diagnose Huntington’s disease.
They refer a person to a neurologist if they suspect that person may have Huntington’s disease.
CT or MRI scans are often recommended by doctors. This can be useful in identifying brain changes and excluding other medical conditions.
Genetic tests are also useful in confirming an exact diagnosis.
Read: Frontotemporal Dementia
Genetic testing
It became possible in 1993 to perform genetic testing on those with Huntington’s disease. You can get genetic testing if you have a family history of the disease.
It is common for people to want a genetic test so they can know if they have the gene and if they are likely to develop symptoms. However, some people would rather not know. Genetic counseling can assist in making this decision.
Huntington’s, genetics and pregnancy
In-vitro fertilization (IVF) is an option for couples who wish to have children and have one of the parents have the gene mutation.
Genetic testing takes place in laboratories, and doctors implant embryos only if the gene mutation has not been detected.
When there is a family history of the condition, genetic testing can also be performed on the fetus during gestation. It is done by taking a placental tissue sample, or chorionic villus, around 10-11 weeks, or by performing an amniocentesis around 14-18 weeks.
Juvenile Huntington’s disease
A doctor will diagnose juvenile Huntington’s disease if symptoms show up before the age of 20.
It can cause leg stiffness, tremors, and regression in learning for children with the juvenile version of the disease.
It usually progresses more rapidly in the juvenile form. Most patients die within the first 10 years after being diagnosed. Complications such as pneumonia or choking are often the cause of death.
Read: Stereotypic Movement Disorder
Huntington disease treatment
There is currently no cure for Huntington’s disease. Its progression cannot be reversed or slowed down by treatment.
Some symptoms can be managed with medication and other therapies.
Medications
Two medications, in particular, have been approved by the Food and Drug Administration (FDA) in order to treat Huntington’s symptoms.
Huntington’s disease can cause jerky, involuntary movements known as chorea that can be treated with tetrabenazine (Xenazine). Suicidal thoughts and actions are also possible side effects.
Besides treating involuntary movements, deutetrabenazine (Austedo) can also treat symptoms of involuntary jaw and tongue movements.
Those who experience mood changes or depression while taking these medications should consult a doctor immediately. It is not recommended for anyone with depression, especially those with suicidal thoughts, to take either medication. If a person needs to avoid these medications for another medical reason, they should consult their physician.
There are a number of other drugs undergoing clinical trials that may help people with Huntington’s disease.
The following drugs may be used in the treatment of movement, outbursts and hallucinations:
- Clonazepam (Klonopin)
- Haloperidol
- Clozapine (Clorazil)
This medication has sedating, stiffening and rigidifying effects.
The following examples of medications may be prescribed for depression and some obsessive-compulsive symptoms:
- Fluoxetine (Prozac, Sarafem)
- Sertraline (Zoloft)
- Citalopram (Celexa)
Read: Facial Tics Disorder
Speech therapy
The muscles of the throat and mouth can be severely affected by Huntington’s disease. The individual has trouble speaking, swallowing and eating because of this.
These skills can be improved and the resulting challenges can be lessened through speech therapy.
Another method of teaching non-verbal people how to communicate without speaking is to use a board with pictures of everyday objects and activities used by non-verbal people.
Physical and occupational therapy
An occupational therapist can help people improve muscle strength and flexibility, which improves balance and reduces the chance of falling.
Exercises can then be designed by the therapist for the patient that fit their level of ability and can be repeated regularly and adjusted as their physical condition changes. The therapist can also provide advice on how to use supporting devices such as a wheelchair or walker
Occupational therapists can provide additional aids that can assist a person with day-to-day functioning, such as handrails in the home, specialized utensils for eating and drinking, and equipment that can help a person get dressed and shower.
Read: Chronic Motor Tic Disorder
Outlook
There are numerous emotional, mental, social and economic effects that Huntington’s disease can have on an individual and their family. The average lifespan for someone with this condition is between 10 and 30 years.
Treatment is currently not possible but can improve people’s quality of life and help them manage the condition.
Scientists hope to find a cure for this condition through gene therapy in the future. Huntington’s disease is a degenerative neurodegenerative disease that has been slowed or prevented by using gene therapy.
According to Emory University scientists, DNA cutting and pasting techniques, such as CRISPR-Cas9, could be of benefit in the future for preventing Huntington’s disease.
Experiments in mice have shown “significant improvements” after 3 weeks. Most traces of the damaging protein had gone, and the nerve cells showed signs of healing themselves.
More research is necessary to determine how healthcare professionals can apply this approach to treat people with Huntington’s disease, but other studies have indicated a similar approach may be successful on a cellular level.
Organizations such as the HDSA help people and their families dealing with the disorder.